

本文讨论的药品审评时间,指申报临床或生产的药品品种从进入药审中心到得出审评结论的时间,这其中包含了每个受理号等待审评的时间,以及技术审评所需花费的时间。
技术审评时间在《药品注册管理办法》中有规定,申报新药临床试验和生产的技术审评工作时间分别为 90 日、150 日,若按工作日来计,分别约为 4 个月、7 个月;160 日的仿制药申请工作时间则约为 7-8 个月。所以,药品审评的大部分时间实际上都花费在等待时间上,该时间则视申报数量和药品审评中心的审评效率等因素来定。
我们选取了 2011-2014 年获得临床和生产批准的 1.1、3.1 类新药以及 2013-2014 年获得批准的 6 类仿制药的具体数据,来一瞥药品审评时间的具体情况。
1.1 类新药的平均审评时间
根据 INSIGHT - China Pharma Data 数据库统计,2011-2014 年获得批准的 1.1 类新药中:
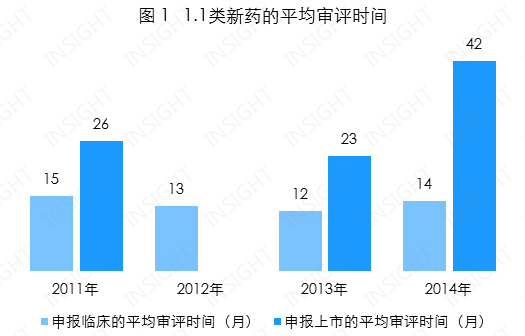
申报临床的平均审评时间为 14 个月,总体趋于稳定;
申报生产的平均审评时间为 29 个月(以获得生产批件为准),2014 年审评时间增加到了 42 个月(见图 1),远超于总体平均水平。
其中,申报上市时间最快的是浙江贝达的盐酸埃克替尼,为 10 个月;最慢的是苏州二叶的阿德福韦酯,历时 64 个月。
另外,江苏恒瑞用于晚期胃癌治疗的甲磺酸阿帕替尼片刚获批不久,其申请上市时间为 38 个月,长于 1.1 类新药申报上市的平均审评时间。该药于 2006 年 4 月申报临床,1 年后获批临床,2011 年申报上市后于 2014 年 10 月 31 日获批。
从这个例子我们可以瞥见 1.1 类新药上市的不易,暂且不论临床前研究的时间,光申报临床到获批上市就历经 8 年之久,真可谓「十年磨一剑」。
中国医药创新论坛会议上的消息称深圳微芯的1.1类新药西达本胺有望以二期临床试验数据获批上市。该药用于复发及难治性外周 T 细胞淋巴瘤(PTCL)的治疗,于 2006 年申报临床,10 个月后获得临床批件,又于 2013 年 3 月申报上市,今年 9 月 25 日完成现场检查,目前 CFDA 数据仍未显示该药的生产批件。
西达本胺若于年底或明年初获批上市,那么其申报上市的审评时间为 22 个月左右,比近 4 年来的平均审评时间提前将近 10 个月,而该药从报临床到获批上市历经将近 6 年。这意味着我国加速创新药品审评的思路在逐步实践中。
3.1 类新药的平均审评时间
INSIGHT 数据库显示,2011-2014 年获得批准的 3.1 类新药中:
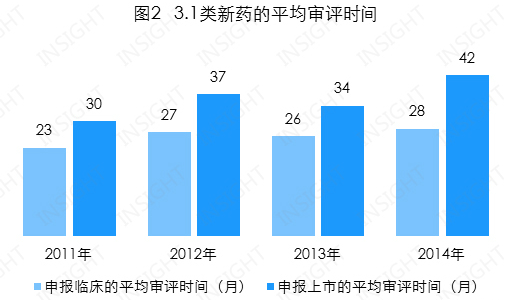
申报临床的平均审评时间为 27 个月,逐年持续增加,总体水平是 1.1 类新药审评时间的近 2 倍;
申报上市的平均审评时间为 34 个月,总体也呈增加趋势,且 2014 年的平均审评时间与 1.1 类新药的审评时间一致,为 42 个月(见图 2)。
近两年,3.1 类申报较为热门的药物当数罗氟司特,该药用于重度慢性阻塞性肺疾病( COPD )的治疗,其原研企业为瑞士奈科明制药公司,分别于 2010、2011 年在欧盟和美国上市,尚未在我国上市。从 2011 年起共有包括奈科明在内的 60 余家企业申报该品种,审评时间的长短即成为了这场「抢仿角逐」中的关键。
目前,合肥立方、重庆华邦、石家庄智恒和山东创新药物研发四个公司获得了临床批件,审评时间在 25-27 个月不等。
3.1 类的抢仿申报像一场马拉松长跑,最终哪家能首先获得生产批件,还需审评时间说了算。
6 类仿制药的平均审评时间
由于 6 类仿制药数据较多,我们仅选取 INSIGHT 数据库中 2013 和 2014 年获得批准的 6 类仿制药进行统计:
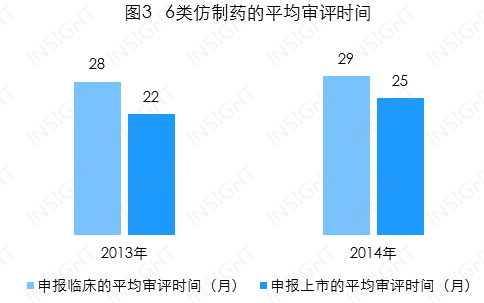
申报临床和申报上市的平均审评时间分别为 29 个月和25 个月,总体均呈小幅上升趋势(见图 3)。
2013 年初国家药监局发布的《关于深化药品审评审批改革进一步鼓励创新的意见》中鼓励探索生物等效性实验的备案制,通过备案后临床试验机构即可以开展试验。
但目前仿制药固体制剂仍然需要先获得生物等效性试验的临床批件才能进行临床试验,该意见尚未实施,并没有发挥出解决仿制药申报积压问题的作用。
除此之外,国家药监局于 2014 年 9 月和 11 月分别发布了两批过度重复上市和申报的药品品种名单,旨在引导企业的研发注册方向,待过度重复申报现象改善,审评速度希望也得以提高。
虽然 2013 年之后申报临床、生产且已获批准的药品,审评时间均少于 20 个月,但 INSIGHT 数据库显示,处于「在审评」状态的 6 类仿制药中,申报临床最早的是在 2011 年 6 月,为河北万岁的盐酸二甲双胍;申报上市最早的是 2010 年 12 月,为重庆市庆余堂的硫酸氢氯吡格雷片。也就是说,部分 2010 年和 2011 年的申报至今仍在排队。
进口药品的审评时间
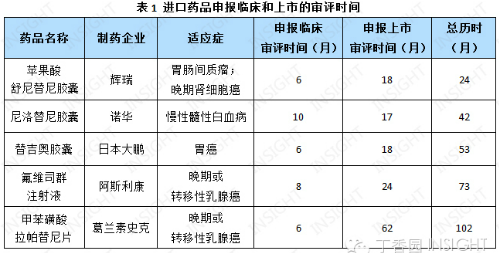
在刚结束的 APEC 工商领导人峰会上,外资制药企业「抱怨」药品审批时间过长。对此,我们从上一篇有关新药获批时间的文章中选取了抗肿瘤领域的 5 个进口新药,根据 INSIGHT 数据库统计这些药品的审评时间:
申请临床的审评时间为 6-10 个月;申请上市的审评时间则跨度较大,快则 20 个月之内,慢则近 24 个月甚至 62 个月。
从总历时来看,平均历时 59 个月,约为 5 年。其中,辉瑞的苹果酸舒尼替尼胶囊审评时间较短,为 24 个月,而葛兰素史克的甲苯磺酸拉帕替尼片从申请临床到获批上市,历经了 102 个月(见表 1),相当于近 9 年的时间。
进口药品如此漫长的审评时间,难怪会引起外资制药企业的「不满」。
由此,「三报三批」话题再一次被热议,丁香园新药与信息讨论版的资深网友 dts1014 也抛出了其观点:
国际多中心临床试验( International MulticenterClinical trial, IMCT )的目的是药物上市,而我国仅将其作为药物研发的一部分来看待,在注册管理办法中写在「药物的临床试验」里,而不是「进口药物的注册和审批」里。
虽然 IMCT 研究所制定的使用剂量、治疗周期、样本量和代表性、体内标志物(比如酶系)、临床试验终点指标以及临床试验所能承受的安全性风险程度,甚至是伦理,不一定能够和中国的临床试验以及临床实践无缝连接,但其作为药物研发的一部分,其最终目标本就是上市。
所以,我们不应该人为的把「药物注册临床申请」和「药物研发临床申请」分离开来,应该从科学和逻辑的角度出发去定义药物的注册程序,而不是人为的割裂它。
另外,「三报三批」体现了 IMCT 申请者和 CFDA/CDE 之间利益的博弈:一边追求全球同步上市,尽快回收研发成本并产生利润;一边考虑药物长期和大范围使用的安全性和有效性。
但是,冲突产生的另一个主要原因是我们没有清晰的法律来定义 IMCT 以及它在中国进口药物注册申报体系中的地位,更加没有类似日本和美国对国际多中心临床试验(美国将其定义为 Multi-regional Clinical trial, MRCT )的指导原则。
因此,「三报三批」不仅反映出审评时限延长的问题,而背后隐藏的问题是我国药物审评审批理念的落后。
综上所述,无论是进口药品,亦或是国内的 1.1、3.1 类新药和 6 类仿制药,药品审评时限过长都已成为必须要面对和解决的问题,因为这个问题大到影响公众用药可及性,小到影响企业的生存。
若要解决此问题,大刀阔斧地改革和一招鲜吃遍天都不太现实,或许可以从最棘手的仿制药审评和验证性临床审评积压等问题着手解决。然而,最重要的是,改变需要魄力,也需要坚持。
盼望药品审评时间的缩短,存在于一个触手可及的未来中。
